长期以来,癌症遗传学和表观遗传学研究人员一直专注于基因和蛋白质的改变,以了解是什么导致细胞癌变和恶性。在细胞核中,高度复杂的基因网络之间存在串扰,由分散的小调节元件通过 DNA 区域的物理接触控制。
许多癌症显示出这种串扰的改变,例如,由于 DNA 的增加、丢失或重排。虽然基因可能非常好,但在细胞核中处于不同位置会破坏它们与调节元件进行物理接触的能力。对这种称为相互作用组的相互作用网络的研究是癌症生物学中最具挑战性和未知的领域之一。
分析相互作用组并非易事。在仔细提取癌细胞的 DNA,采取一切可能的预防措施避免破坏基因组中“相互交谈”的那些部分后,科学家们使用一些生化技巧将它们粘在一起,以揭示哪些基因和调控元件处于物理状态接触。通过将健康细胞与癌细胞进行比较,研究人员希望了解恶性肿瘤的异常“话语”。
到目前为止,这种方法需要大量的 DNA 才能成功,因此,由于活检中获得的样本量有限,因此在临床环境中没有用处。但由于 liCHi-C,这种情况可能会改变——一种由 3D 染色质组织实验室开发的新方法,该实验室由 Josep Carreras 白血病研究所的 Biola M. Javierre 博士领导,
在Nature Communications最近发表的一篇文章中,研究人员表明,可以直接从患者样本中分析特定肿瘤的相互作用组,而不是从体外模型中分析,这是一个巨大的飞跃。这一壮举依赖于以下事实:liCHi-C 是对之前的启动子捕获 Hi-C (PCHi-C) 方法的改进,仅适用于 50,000 个细胞,而不是其他方法所需的数百万个细胞。由于简化的实验方案与新的生物信息学工具相结合,样本量的大幅减少成为可能。
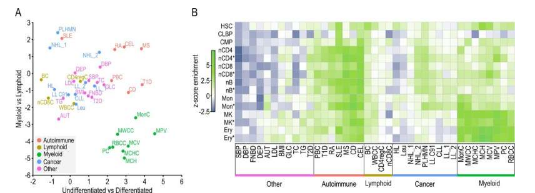
与 Josep Carreras 研究所、巴塞罗那超级计算中心、Wellcome-MRC 剑桥干细胞研究所、Sant Joan de Déu、IDIBAPS 和 Hospital Clinic 等机构的同事一起,研究人员已经能够在开发过程中提高相互作用组图的分辨率来自健康捐赠者和癌症患者的造血干细胞,使他们能够识别白血病中发生的改变网络。此外,他们还展示了如何使用 liCHi-C 以更高的精度识别大型 DNA 重排,并揭示非编码改变在癌症发展中的作用。