特拉维夫大学萨克勒医学院和萨戈尔神经科学学院的一个研究小组首次发现了导致神经退行性疾病 ALS 神经破坏的生物学机制。由 Eran Perlson 教授和博士生 Topaz Altman 和 Ariel Ionescu 领导的开创性研究表明,这种致命疾病的病程可以在早期阶段延缓甚至逆转。它是与 Sheba 医疗中心神经肌肉疾病诊所主任 Amir Dori 博士合作进行的。研究结果发表在《自然通讯》上。
ALS 是最常见的运动神经元疾病类型,会导致瘫痪和肌肉萎缩。每 400 人中就有 1 人患有这种疾病,但目前尚无有效的治疗方法。ALS患者逐渐失去控制随意肌肉运动的能力,导致完全瘫痪,最终失去独立呼吸的能力。ALS患者的平均预期寿命目前只有三年左右。
Perlson 教授解释说:“直到今天,还不清楚是什么导致了这种疾病”。“只有大约 10% 的患者具有已知基因突变的家族背景,但其余 90% 是一个谜。这种疾病引起的瘫痪是由于运动神经元受损,导致神经末梢退化和肌肉神经支配的丧失。这最终导致神经退化和脊髓运动神经元死亡,但是直到现在我们还无法理解导致这种恶性级联反应背后初始损伤的基本生物学机制。
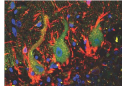
实验中使用的模型动物神经肌肉接头的代表性图像。运动神经为绿色,肌肉纤维上的感受器为紫色。右边的照片是在低倍率 (100x) 下拍摄的,而左边的照片是在高倍率 (600x) 下拍摄的。图片来源:特拉维夫大学
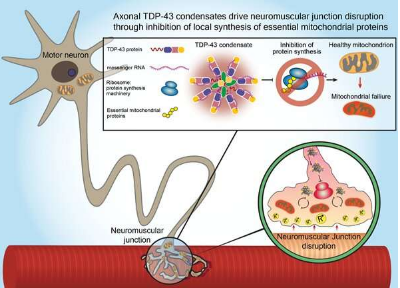
为解开这个谜团,特拉维夫大学的研究人员专注于一种名为 TDP-43 的蛋白质,早期研究表明,这种蛋白质在大约 95% 的肌萎缩侧索硬化患者的大脑中以不同寻常的数量和定位聚集。Perlson 教授和他的团队揭示了蛋白质积累与运动神经元末梢和肌肉之间的突触退化之间的一种新的生物学联系,称为神经肌肉接头,它将神经指令转化为身体运动。在肌萎缩侧索硬化患者的肌肉活组织检查中,研究人员发现,在疾病的早期阶段和患者出现任何严重症状之前,有毒蛋白质也在这些神经肌肉接头附近积聚。在研究人员进行的一系列实验中,在 ALS 患者和转基因模型动物的细胞中,他们发现 TDP-43 蛋白在神经肌肉接头中的积累抑制了局部合成对线粒体活动至关重要的蛋白质的能力,这为基本细胞过程提供了动力. 神经末梢线粒体功能障碍导致神经肌肉接头破坏,最终导致运动神经元死亡。“首先了解运动神经元的空间复杂性很重要”,Perlson 教授说。神经末梢线粒体功能障碍导致神经肌肉接头破坏,最终导致运动神经元死亡。“首先了解运动神经元的空间复杂性很重要”,Perlson 教授说。神经末梢线粒体功能障碍导致神经肌肉接头破坏,最终导致运动神经元死亡。“首先了解运动神经元的空间复杂性很重要”,Perlson 教授说。
我们表明,在 ALS 中,这种蛋白质离开细胞核并在整个细胞中积累,尤其是在神经肌肉接头中。由于运动神经元的功能取决于位于“延长线”远端的这些神经肌肉接头,我们意识到这一发现可能至关重要。我们发现 TDP-43 蛋白在神经肌肉接头处形成的积聚会捕获 RNA 分子并阻止线粒体功能必需蛋白质的合成。线粒体是在细胞中发现的细胞器,是许多细胞过程(包括神经传递)的主要能量提供者。TDP-43 蛋白在神经肌肉接头处的凝结导致严重的能量消耗,阻止线粒体修复,脊髓中的运动神经元。”
为了证实他们的发现,特拉维夫大学的研究人员决定使用美国一组研究人员最近发表的一种实验分子,用于开发另一个目的——通过分解神经延伸中的蛋白质凝聚物来增强损伤后的神经再生。研究人员证明,这种分子还可以分解 ALS 患者细胞中的轴突 TDP-43 蛋白凝聚物,这一过程提高了产生必需蛋白的能力,增强了线粒体活性,并防止了神经肌肉接头退化。此外,在模型动物中,研究人员表明,通过逆转 TDP-43 在神经和神经肌肉接头中的积累,能够恢复退化的神经肌肉接头,并使患病的模型动物几乎完全康复。